
Pharmacogenetics Studies in STAR*D: Strengths, Limitations, and Results
Major depression is a severe medical condition that is predicted to be the second leading cause of death and disability worldwide by the year 2020 ( 1 ). Few predictors of response to pharmaco- and psychotherapeutic treatments are known ( 2 ), and finding the most effective treatment for each patient is difficult. Response rates for any single medication are about 50%. For only a minority of patients do symptoms fully remit after about six months of treatment and sometimes with trials of two or more medications ( 3 ). Tolerability and adverse events are common factors contributing to nonadherence and treatment discontinuation, which result in persisting symptoms and disability. In addition, concerns about rare but severe side effects, such as treatment-emergent suicidal ideation, may reduce access to and acceptability of treatment ( 4 ).
These facts suggest that a better understanding of the pathophysiology of depression and identification of useful biological markers, including genetic markers, could improve treatment selection and help match treatment to desirable clinical outcomes, leading to new therapeutic strategies, drug targets, and conceptualizations of depression. For example, genetic markers may help predict antidepressant treatment response or adverse events; however, few clinical applications have resulted ( 5 ).
Technological advances in human genetics over the past decade, along with the availability of large treatment cohorts for study, such as in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) ( 6 ), promise to transform pharmacogenetics into one of the foundations of evidence-based medicine. This article reviews the implications of STAR*D findings for pharmacogenetics research.
What did STAR*D contribute to pharmacogenetics research?
Two major developments in recent years that have begun to move the field of pharmacogenetics forward are the availability of large, well-characterized samples and the advent of genomic technologies of unprecedented power and efficiency. The role of STAR*D has been pivotal in the advancement of the pharmacogenetics of antidepressant response and adverse effects. STAR*D provided for the first time a sample of well-characterized, prospectively followed participants large enough to detect even modest genetic effects. The genetic studies conducted to date include investigations of single-nucleotide polymorphisms (SNPs) in multiple candidate genes. In the serotonin transporter gene (SLC6A4), the 43 base pair promoter-linked insertion-deletion region (LPR) and the variable-number tandem repeat (VNTR) polymorphism in the second intron have also been studied. In addition, a preliminary genomewide association study (GWAS) has been performed with selected participants, and a GWAS of the full set of participants who provided DNA is under way.
Sample characteristics
The STAR*D centers enrolled a clinically representative cohort of about 4,000 adults with nonpsychotic major depression. All were treated with citalopram at level 1 for up to 12 weeks and evaluated prospectively for treatment response and adverse events. About 2,000 individuals were asked to provide a DNA sample—most of whom agreed—but participants who dropped out early are underrepresented among the available DNA samples (see below).
Large samples are a key element of robust findings in genetic and pharmacogenetics studies ( 7 ), and STAR*D represents an order-of-magnitude increase over the size of all previously studied samples with major depression. An added advantage of large samples is that without complete loss of statistical power, they allow for stratification and inclusion of covariates that might represent important confounders.
The intent of STAR*D investigators was to enroll a cohort of outpatients that resembled those seen at primary and specialty care clinics around the United States, including a typical mix of race, ethnicity, and socioeconomic status. Therefore, few exclusion criteria were used, and participants with medical conditions or anxiety disorders (except primary obsessive-compulsive disorder) were included in the trial. STAR*D had no placebo arm; thus all participants received active treatment. In the overall sample, over 70% of participants had experienced two or more prior episodes of depression or a sustained episode during the two years before study entry, and about 60% had one or more comorbid psychiatric disorders ( 3 ).
Treatment was organized into four levels. Each offered a number of treatment options for up to 12 weeks. All participants began with 10–60 mg per day of citalopram as tolerated (first step or level 1). Participants who did not respond sufficiently could choose random assignment to level 2, which included options for switching to any of three other antidepressants (sertraline, bupropion, or venlafaxine) or to cognitive therapy or for augmenting citalopram with either of two other medications (bupropion or buspirone) or with cognitive therapy. For level 2 participants who did not respond sufficiently, random assignment to level 3 included two medication switch options (mirtazapine or nortriptyline) and two medication augmentation options (lithium or thyroid hormone). Level 4 included one of two switch options (tranylcypromine or the combination of mirtazapine and venlafaxine).
Participants at any level who responded sufficiently to treatment were transitioned to follow-up status for up to 12 months. DNA samples were available from 1,914 level 1 participants, 883 level 2 participants, and 255 level 3 participants and from the 85 participants who continued to level 4.
The various pharmacological and nonpharmacological approaches used could potentially provide information on antidepressant outcomes and tolerability of the other therapeutic agents; however, the samples may not be large enough to screen for alleles of small effect.
Power
The large size (N=1,914) of the uniformly treated sample is crucial to obtain the necessary power to detect differences in treatment outcome or emergence of adverse events. Relatively low effect sizes can be detected even in the presence of rare phenotypes, such as treatment-emergent suicidal ideation (STAR*D prevalence 6.3%), and markers with low (<10%) minor allele frequencies, such as those described in GRIK2 ( 8 ) (see below).
Diversity
Few, large, well-characterized samples of patients treated with antidepressants are available for study along with their DNA. The availability of samples from racial-ethnic minority groups is significantly more problematic. STAR*D included all patients regardless of race and ethnicity. This provided both an opportunity to study racial-ethnic differences in treatment response and side effects and the option of analyzing these groups as separate cohorts. STAR*D had a subsample of 313 self-reported African-American participants; however, the sample may not be large enough to provide the statistical power to study genetic variants with small effects or low allelic frequency. This is also the case for the Hispanic subgroup of 274 participants (eight black and 247 white Hispanic participants [19 mixed race]). Other racial-ethnic groups were represented by only small numbers of participants, including 21 Asians, 16 Native Americans, eight Pacific Islanders, and a cohort of 68 "multiracial" participants (all self-reported). The largest group included 1,279 white, non-Hispanic participants.
As noted, a group of individuals with a variety of racial-ethnic backgrounds should be analyzed carefully, with consideration of potential confounders, such as racial stratification. Current methods to address these issues include calculating posterior probabilities of racial-ethnic group membership derived from genotypes obtained from the sample by use of publicly available software such as STRUCTURE ( 9 , 10 ) or EIGENSTRAT ( 11 ). These values can be used as covariates in the association equation to "weight" racial-ethnic background in the overall result.
Prospective assessment of outcome and side effects
Participants in the STAR*D trial were assessed at baseline and about every two weeks thereafter for up to 12 weeks at each level. The instruments used to assess outcome were the 17-item Hamilton Depression Rating Scale (HAM-D) ( 12 , 13 ) and the clinician-rated and self-rated 16-item Quick Inventory of Depressive Symptomatology (QIDS-C and QIDS-SR) ( 14 ). The baseline visit also included the 30-item Inventory of Depressive Symptomatology ( 15 ). The HAM-D was used only at baseline and each level endpoint. Both the QIDS-C and QIDS-SR were completed at every visit. In addition, all postbaseline visits included a systematic assessment of adverse effects that used the Frequency, Intensity and Burden of Side Effects Rating and the Global Report of Side Effect Burden ( 16 ). Although a detailed discussion of the instruments used in STAR*D is beyond the scope of this article, these instruments have been widely cross-validated and had a good interrater reliability ( 17 ). Participants were followed closely, allowing for multiple datapoints in regard to ongoing symptoms and experience of side effects, which minimized recall bias.
Treatment
Another significant advantage of STAR*D is that all participants were treated with citalopram at level 1. This highly selective serotonin reuptake inhibitor (SSRI) was chosen because of its proven efficacy in major depressive disorder, good tolerability profile, and low potential for pharmacokinetic interactions with other medications, particularly those used by patients with general medical illnesses. Although treatment with a single drug limits potential generalizability of findings to other antidepressants, it has the advantage of having fewer pharmacokinetic and pharmacodynamic confounders for assessment of adverse effects and outcome. It is important to note that one of the general principles in STAR*D was to ensure that participants on all regimens had an adequate treatment trial of at least four to eight weeks of drug administration at recommended dose ranges.
As noted, citalopram nonresponders were offered options to switch or augment their ongoing treatment. At level 2, participants augmented their citalopram treatment with bupropion (N=184), buspirone (N=177), or cognitive therapy (N=63) or switched to other drugs, including bupropion (N=135), sertraline (N=139), and venlafaxine (N=154), or to cognitive therapy (N=31). At level 3 patients could be switched to mirtazapine (N=74) or nortriptyline (N=80) or have their ongoing treatment augmented with lithium (N=30) or thyroxine (N=33). At level 4, the options were limited to tranycypromine (N=45) or a venlafaxine-mirtazapine combination (N=40).
In levels 2 through 4 pharmacogenetics studies of outcome and analyses of side effects are complicated by the fact that the samples are small; however, exposure to other antidepressants implies little or no response to the previous regimen, which suggests that there is a subgroup of patients who do not respond to a variety of pharmacological interventions.
What are the limitations of STAR*D as a pharmacogenetics study?
Perhaps the greatest advantage of STAR*D from a clinical perspective is also its greatest limitation for study of pharmacogenetics. Specifically, as an effectiveness study, STAR*D by design included patients with substantial medical and psychiatric comorbidity. Because participants were drawn from primary as well as specialty care settings, the general medical burden exceeds that of a typical clinical trial ( 18 ). Participants could also receive concomitant treatment with medications, such as beta blockers, that could have an impact on mood or treatment response. Similarly, individuals with ongoing substance misuse were eligible for participation as long as they did not require additional treatment targeting their co-occurring conditions ( 19 , 20 ). Consistent with other clinical trials, STAR*D excluded patients with severe suicidality and limited the sample to nonpsychotic outpatients. Although the broad inclusion criteria greatly increase the generalizability of STAR*D findings to clinical practice, they also introduce heterogeneity that may make genetic effects more difficult to detect. On the other hand, if one goal of pharmacogenetic testing is to develop clinically useful diagnostic tools, clinically representative populations are precisely the ones that require study.
Several other features of STAR*D may increase sample heterogeneity and thereby diminish power to detect genetic associations. First, STAR*D did not include detailed assessments of medication adherence at level 1 (citalopram), such as pill counts or measurement of blood citalopram levels. Failing to consider nonadherence could lead to misclassification of outcomes (for example, when poorer outcome is a result of treatment nonadherence). This concern is likely to be more than theoretical, because adherence to treatment with antidepressants is known to be poor in general practice ( 21 ). Second, although most of the relevant sociodemographic characteristics were ascertained, such as gender, race-ethnicity, income and employment, and marital status, others, such as social support or religiosity, that have been previously linked to antidepressant outcome were not included in STAR*D ( 22 ). Personality disorders have been reported as potential confounders of antidepressant treatment outcome in some studies, including recent meta-analyses ( 23 ), but not all ( 24 , 25 ). STAR*D did not assess personality traits; however, it is important to note the well-known inaccuracy of assessment of trait characteristics, such as personality disorders, in the context of depressive states ( 26 ).
Another STAR*D limitation is the absence of a placebo arm at level 1. As a result, participants classified as treatment responders can be considered as two admixed populations: those whose improvement was attributable to citalopram and those whose improvement was attributable to placebo. (In fact, the placebo literature suggests that the latter group can be subdivided further—for example, by identifying those whose improvement is attributable to time or regression to the mean and those whose improvement is a result of the placebo effect.) A very simplistic response to this aspect of the design is to raise concern about the specificity of any associations: for example, might variation in genes associated with treatment response simply be linked to shorter episodes of depression regardless of intervention? It can also be argued that specificity is a second-order question—that is, "after" finding effects one could then proceed to determine specificity in other data sets. Indeed, this next step would be necessary even with a placebo arm, because there was also no active comparator at level 1.
It also bears emphasis that strategies exist to partially address the problem of placebo response. One approach applied in clinical investigations is to examine "patterns" of response that are characteristic of "true-drug" response and patterns characteristic of placebo response ( 27 , 28 , 29 ), although more recent data cast some doubt on the utility of these parameters ( 30 ). Alternatively, one might focus on response to next-step treatments (that is, level 2 and subsequent levels), presuming that placebo response should be greatly diminished after an initial treatment failure ( 31 ).
One further limitation in the STAR*D genetic data set is the difference between individuals who participated in the genetics study and the STAR*D cohort as a whole, which is discussed in detail elsewhere ( 32 ). Because the genetics portion of the study was added after study initiation, participants in that portion would be skewed toward those who entered the study later (which should not introduce bias) and those who remained in the study longer (either because of good treatment response and participation in follow-up or because of poor treatment response leading to continued participation in subsequent levels (which could well introduce bias). Moreover, a substantial literature documents that ethnic and racial groups may differ in their willingness to participate in genetics studies ( 33 ). In general, although these distinctions should have little impact on the detection of associations in most cases, they could certainly have an impact on the generalizability of results.
Biomarkers of depression treatment outcome have been scarce. Thus there was no justification for collecting serum or whole blood in STAR*D. However, new and more sophisticated techniques may arise that could provide important information about these phenotypes. The lack of these biological materials for STAR*D participants may limit associations between genetic variations and their function.
Finally, STAR*D has some limitations for investigation of tolerability outcomes that bear consideration. In particular, the primary measure of adverse effects by bodily system—the Patient-Rated Inventory of Side Effects—was not administered at study entry. Therefore, subsequent reports of adverse effects cannot be distinguished from preexisting symptoms. This consideration was apparent in analyses of sexual symptoms, where it was impossible to determine whether these symptoms were truly treatment emergent ( 34 ). To circumvent this problem, one approach is to consider only adverse effects not present at the initial postbaseline visit. Alternatively, some potential adverse effects, such as insomnia, can be identified by using items on ratings scales that were completed at baseline (Laje G et al., unpublished data).
Results of pharmacogenetics studies in STAR*D
The STAR*D study aimed at helping clinicians make treatment decisions by elucidating which options provided better efficacy and tolerability. STAR*D is the largest pharmacogenetics study of major depression. It has provided, and continues to provide, a tremendous opportunity to elucidate genetic determinants of treatment outcome and tolerability.
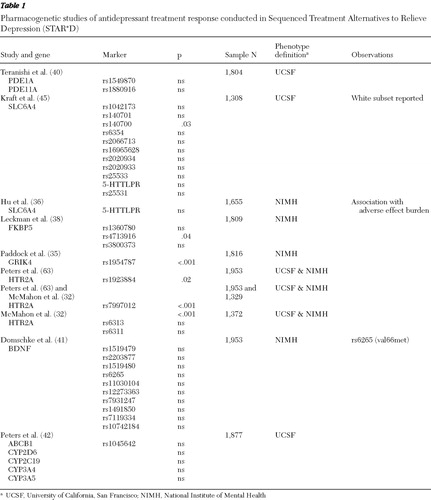
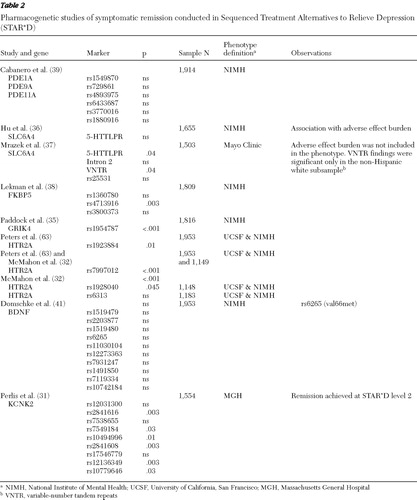
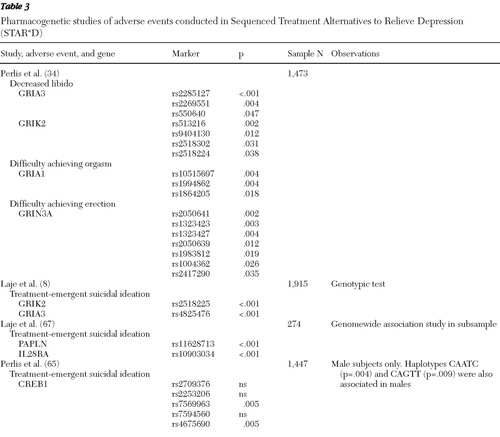
Two common genetic analytic approaches have been used in the STAR*D sample: candidate gene and genomewide association studies. We discuss results of candidate gene studies and describe the genomewide studies that have been conducted in a subsample of participants. The genomewide results for the whole sample are not yet published. Two general phenotype groups have been studied: antidepressant treatment outcome and treatment-emergent adverse effects ( Tables 1 , 2 , and 3 ).
 |
 |
 |
Antidepressant outcome
The STAR*D sample offered an unparalleled opportunity to search for new genes linked to antidepressant outcome and to replicate previous findings. Novel associations with citalopram response were reported within HTR2A (the gene that encodes the serotonin 2A receptor) ( 32 ) and GRIK4 (encodes the KA1 subunit of the glutamate-kainate receptor) ( 35 ). The KCNK2 gene (encodes a potassium channel of the K subfamily) was associated with treatment resistance ( 31 ).
The serotonin transporter (SLC6A4) is a gene for which new variants have been described and replication of previous findings have been attempted ( 36 , 37 ). Other genes of interest that were previously reported to be significant included FKBP5 ( 38 ), PDE11A ( 39 , 40 ), and BDNF ( 41 ). Finally, genes with pharmacokinetic function were also assessed in this group and included CYP2D6, CYP2C19, CYP3A4, CYP3A5, and ABCB1 ( 42 ).
Although these reports use an outcome phenotype, the phenotype definition varies across groups (see below). This is particularly problematic when analyzing genes such as the serotonin transporter (SLC6A4) because tolerability is a confounder of outcome and the results could vary depending on whether it is included. This is particularly the case when vigorous dosing is used; that is, the nonresponding patient receives ever-increasing doses until intolerance is encountered or the intolerant patient cannot tolerate a therapeutic dosage.
Response and remission phenotype definitions
Four groups across the United States have tested outcome phenotypes in the STAR*D sample. Although the outcomes were antidepressant treatment response and remission, all the phenotype definitions have subtle yet significant differences.
The Intramural Research Program at the National Institute of Mental Health (NIMH), led by one of the authors (FJM) in close collaboration with another author (AJR) and others, crafted a phenotype based on stringent criteria for time taking the drug (six-week minimum), a HAM-D score of >10 at baseline, treatment adherence (by report), tolerability, and at least 50% improvement in QIDS-C (clinician-rated) scores for "response" or a score of ≤5 on the QIDS-C at endpoint for "remission." A third phenotype, QIDS-C change, was defined on the basis of the same exclusions described above. This phenotype used the relative change in QIDS-C score between baseline and endpoint to define a percentage of improvement or worsening. The goal for such stringent phenotype definition was to guarantee, to the extent possible, that nonresponders would have had a fair chance to respond to the drug, achieving at least six weeks of treatment at a therapeutic dosage ( 32 ).
A group at Massachusetts General Hospital headed by another author (RHP) used definitions similar to those of the NIMH group but also examined outcomes without considering tolerability.
A group at the University of California, San Francisco (UCSF), led by Steven P. Hamilton, defined six interrelated phenotypes. On the basis of exposure to the drug (at least six weeks), patients were considered responders if they had a reduction of 50% or greater in QIDS-SR (self-rated) scores and nonresponders if the score had decreased less than 50% at the level 1 endpoint. Remitters were defined by using the same time criterion and a QIDS-SR score of <6. The UCSF group also defined a "specific responder" phenotype, in which the level of improvement needed to qualify as a responder was sustained after all remaining visits. The intent was to weed out possible placebo response. Finally, a tolerance outcome was based on study exit data. All participants who continued with citalopram were considered tolerant, and patients who refused to continue level 1 treatment or who left the study because of side effects were considered intolerant ( 42 ).
A group at the Mayo Clinic, led by David A. Mrazek defined three outcomes—remission, response, and citalopram tolerability—but reported only the definition and the results for remission. Remission was defined as a score of <6 on the QIDS-C at the last clinic visit. In addition, participants needed a baseline QIDS-C score of ≥10 or greater, at least six weeks of treatment, and citalopram adherence ( 37 ).
The HTR2A and GRIK4 genes
The first pharmacogenetics results from STAR*D were reported by McMahon and colleagues in 2006 ( 32 ), who sampled 768 SNPs in 68 candidate genes. To address the issue of multiple testing, a split-sample design was used. The criteria for an association to be considered significant were same marker, same allele, and same test in both test and replication subsamples. Three phenotypes were tested: response, remission, and QIDS-C change from baseline to endpoint as described above (NIMH phenotype definitions). Results of the primary analysis showed two associated markers: one located in the second intron of the HTR2A gene (rs7997012) and the other located in the GRIK4 gene (rs1954787) ( 32 , 35 ) ( Tables 1 and 2 ). The homozygous carriers of both the HTR2A and GRIK4 response-associated alleles were 23% less likely to be citalopram nonresponders than participants with none of these marker alleles. The area under the curve expressing the probability of correctly identifying a responder from a random pair of participants was .58, suggesting that these two markers have very small predictive value ( 35 ). These findings have yet to be replicated in an independent sample. However, positron emission tomography studies using [ 11 C]DASB, a selective tracer for measuring in vivo serotonin transporter density, have found associations with rs7997012 (HTR2A) in thalamus and rs1954787 (GRIK4) in the pregenual cingulate cortex, suggesting that these two variants might have a subtle regulatory effect on the serotonin transporter and thus influence antidepressant treatment through this mechanism (Laje G, Cannon DM, Allen AS, et al., unpublished manuscripts, 2009).
The serotonin transporter
The serotonin transporter (SLC6A4) and a functional polymorphism in its promoter region (known as serotonin transporter linked polymorphic region: 5-HTTLPR) have received the most attention by mental health researchers. As the proximal target for SSRIs, the most widely prescribed antidepressants, this gene is the most obvious pharmacodynamic candidate for association studies. This variant, the insertion of a 43 base pair segment in the promoter region (L-allele) together with the A-allele of the SNP rs25531 in the same region seem to affect gene expression in important ways ( 43 ), and thus differential expression of the SSRIs' target became a good hypothesis to explain SSRI efficacy. A meta-analysis of 15 published studies that included 1,520 patients concluded that there was evidence in favor of a significant association of the long (L) allele with better response to SSRIs ( 44 ). This result may reflect publication bias because negative studies tend not to get published.
The serotonin transporter was widely studied in STAR*D. Three publications reported results of SLC6A4 variation and treatment outcome ( 36 , 37 , 45 ) ( Tables 1 and 2 ). Kraft and colleagues ( 45 ) reported no association between citalopram treatment response and nine tagging SNPs and the LPR. This study used the UCSF phenotype definitions. Hu and colleagues ( 36 ), using the NIMH outcome definitions, found that LPR variation, specifically the L A allele, was associated with tolerability and not with treatment outcome, and thus carriers for the short (S/deleted) or the less functional L G alleles had an increase in side-effect burden that could lead to discontinuation and nonresponse. Finally, Mrazek and colleagues ( 37 ) conducted analogous experiments but also selected four subsamples (N=60 each) of self-described Caucasians, African Americans, Asians, and Mexican Americans (N=240) from the Coriell Cell Repository and sequenced the entire SLC6A4 gene, reporting some novel variants that were later tested in STAR*D. This study concluded that for white non-Hispanic participants there was an association between remission of depression treated with citalopram and both the VNTRs 12/12 genotype in intron 2 and the LPR variant. Although this study's phenotype definition was close to the NIMH remission phenotype, it did not include tolerability. This study specifically excluded 61 participants with known treatment nonadherence, but they may not necessarily have had tolerability issues.
The FKBP5 gene
The FKBP5 gene encodes a chaperone protein important for fine tuning of the hypothalamic-pituitary-adrenal axis (HPA axis). An association between variants in this gene and antidepressant treatment and recurrence of major depressive disorder was first described by Binder and associates ( 46 ). This gene departed from the monoamine realm but remains a good candidate on the basis of its relevance in pathways such as the HPA axis. Lekman and colleagues ( 38 ) conducted a replication study in the STAR*D sample and found the modest effect previously described in rs1360780 and proposed rs4713916 as a putative functional region ( Tables 1 > and 2 ). A subsequently published study has replicated the rs1360780 association in a small cohort of German patients ( 47 ); however, this variant was negative in a Han Chinese population ( 48 ).
The phosphodiesterase genes
In 2006 Wong and colleagues ( 49 ) published a report linking variation in PDE1A and PDE11A to antidepressant treatment outcome in a Mexican-American population (N=284). The PDE1A and PDE11A are two genes from the large phosphodiesterase (PDE) family. These genes are reasonable candidates because they metabolize cyclic adenosine and guanosine monophosphate (cAMP and cGMP). Cyclic AMP also binds to a response-element binding protein (CREB) in the nucleus and regulates gene transcription of brain-derived neurotrophic factor (BDNF) among others ( 50 ).
Two replication attempts were conducted with STAR*D data, the first one by Teranishi and colleagues ( 40 ) and the second by Cabanero and colleagues ( 39 ). The first group evaluated two markers, rs1549870 (PDE1A) and rs1880916 (PDE11A), using a response phenotype based on at least six weeks of treatment and >50% improvement in QIDS-SR score for response and a QIDS-SR score <6 for remission with Hispanic, Caucasian, and African-American subsamples (see UCSF definitions) ( Table 1 ). Cabanero and colleagues analyzed the Hispanic subsample (N=268) and the entire cohort (N=1,914) ( Table 2 ). Among other markers, those previously reported on PDE11A (rs1880916), PDE1A (rs1549870), and PDE9A (rs729861) were analyzed for association with treatment outcome by using both a phenotype similar to that used by Wong and colleagues and the NIMH outcome definition. Neither report found evidence of association with markers in PDE11A, PDE9A, or PDE1A on individual or haplotype tests in the Hispanic, Caucasian, or African-American subsamples or the entire cohort. However, in the study by Cabanero and colleagues, one of the markers—(rs4893975) in PDE11A—was found to be in Hardy-Weinberg disequilibrium in cases of depression, suggesting that this gene might have a role in major depressive disorder but not in antidepressant response.
The BDNF gene
The BDNF gene is a member of the neurotrophin family and is involved in neuronal growth and plasticity. Variation in BDNF has been thought to play a role in the etiology of affective disorders and the mediation of antidepressant treatment response ( 51 ). Furthermore, expression of BDNF has been shown to be modified by antidepressant treatment ( 52 ). A functional nonsynonymous SNP causing an amino acid substitution of valine to methionine has been identified in codon 66 (val66met; rs6265) ( 53 , 54 ).
Multiple studies support a potential role of BDNF variation in the pathogenesis of major depressive disorder ( 55 , 56 , 57 ), but not all studies have done so ( 58 , 59 ), including a recent meta-analysis ( 60 ). The BDNF val66met polymorphism has been at the center of antidepressant treatment response with some positive ( 61 ) and negative ( 62 ) results.
A study by Domschke and colleagues ( 41 ) found no evidence of an association between treatment outcome and BDNF variation, including the val66met, in a German sample (N=268) or the STAR*D sample (N=1,914) ( Tables 1 and 2 ).
Genes involved in pharmacokinetics of antidepressants
Genes that regulate the bioavailability of antidepressant drugs could also have a significant impact on response and tolerability. The majority of antidepressant compounds are metabolized by the liver through the cytochrome P-450 pathway and its enzymatic subgroups. Genes such as CYP2D6, CYP1A2, CYP3A4, and CYP2C19 have been studied and well characterized, but their clinical use has proven to be limited. The ABCB1, another pharmacokinetic gene, also known as MDR1 (multidrug resistance) encodes the p-glycoprotein and is expressed in tissues that have elimination or protective roles, such as the intestines, liver, kidney, and blood-brain barrier. Peters and colleagues ( 42 ) published a study using a split-sample design to assess 15 polymorphisms from five pharmacokinetic genes, including CYP2D6, CYP2C19, CYP3A4, CYP3A5, and ABCB1. Using the UCSF phenotype definitions, they found no association with citalopram outcome in any of these genes ( Table 1 ).
The KCNK2 (TREK1) gene
On the basis of rodent models of antidepressant response, Perlis and colleagues ( 31 ) examined variants in four genes—KCNK2 (TREK1), SLC18A2 (VMAT2), S100A10, and HDAC5—for association with treatment resistance in STAR*D, which they defined as not achieving remission despite two adequately tolerated treatment trials (that is, levels 1 and 2). The remission phenotype was defined as a QIDS-C score <6. Although no association with any of the four genes was found at level 1 alone, in the primary analysis of treatment resistance, variants in KCNK2 were associated with treatment response among patients with unsatisfactory response to both citalopram and one additional treatment level ( Table 2 ). These findings suggest that genetic variation in KCNK2 may have a role in treatment resistance, but they suggest more broadly that animal models might be used in pharmacogenetics studies of antidepressant response.
Other genes
The initial study by McMahon and colleagues ( 32 ) sampled 68 candidate genes with 768 markers. These genes represented major neurotransmitter systems involved in depression. Marker coverage was limited by financial resources, and gene priority was established by an expert panel. Briefly, the sampled genes were serotonin related (HTR1A, 1B, 1D, 1E, 1F, 2A, 2B, 2C, 3A, 3B, 3C, 3D, 3E, 4, 5A, 6, 7, SLC6A4, TPH1, and TPH2), norepinephrine related (DBH and SLC6A2), glutamate ionotropic receptor and transporter genes (GRIN1, 2A, 2B, 2C, 2D, 3A; GRIA1-4, GRIK1-5, and SLC1A1), dopamine and neutrophic factors (COMT, TH, MAOA, BCL2, BDNF, and NTRK2), and other genes known to be relevant in depression (see reference 32 , Table 2 , for a comprehensive list).
Because of the number and size of these genes, some were not exhaustively covered. Therefore, although all of them were included in the outcome phenotype analyses, variation not sampled may still prove to be important to this phenotype. The findings imply a preliminary negative association of these genes and antidepressant outcome, but given the limited coverage the study could not rule out the presence of markers that might have some relevance to these phenotypes.
Peters and colleagues ( 63 ) selected five serotonin-related genes and resequenced the exonic and putatively regulatory regions of HTR1A, HTR2A, TPH1, TPH2, and MAOA in a fluoxetine-treated sample. These investigators uncovered some novel variants that were not associated with response to citalopram in STAR*D when the UCSF phenotype definitions were used.
Adverse effects
The emergence of adverse reactions is common to all pharmacological treatments. The severity of these effects, however, determines whether a patient will be partially adherent, continue, or discontinue treatment. Finding genetic predictors has been a priority for many groups, especially predictors of adverse events that would interfere with treatment because of their severity or risk. Two phenotypes have been studied in STAR*D: treatment-emergent suicidal ideation and sexual dysfunction ( Table 3 ).
Treatment-emergent suicidal ideation
Suicidal ideation is an uncommon symptom than can emerge during antidepressant treatment. The Food and Drug Administration determined after a meta-analysis that there was sufficient evidence to issue a "black-box warning" highlighting the risk of treatment-emergent suicidality among patients taking antidepressants who were under age 25 ( 64 ). To address treatment-emergent suicidal ideation, two candidate gene studies and one genomewide study were conducted with STAR*D data.
First, in a candidate gene study of the cyclic adenosine monophosphate response-element binding protein (CREB1), Perlis and colleagues ( 65 ) reported two SNPs and two of the five SNP haplotypes associated with treatment-emergent suicidal ideation among men. The phenotype was defined as a QIDS-C score ≥2 on item 12 at any postbaseline visit for participants whose baseline score was 0 or 1. Subsequently, Laje and colleagues ( 8 ) used a different phenotype definition based on item 12 of the QIDS-SR, in which participants whose baseline score was 0 had a score ≥1 on any postbaseline visit. These investigators reported results from the 68 candidate gene data set from the NIMH group. In this study, two markers—one in GRIA3 (rs4825476) and another in GRIK2 (rs2518224)—were associated with treatment-emergent suicidal ideation. Different alleles on GRIA3 and GRIK2 were also implicated with treatment-emergent suicidal ideation in an independent sample of German inpatients with major depressive disorder in which the phenotype was derived from item 3 of the HAM-D ( 66 ).
Finally, the NIMH group conducted a genomewide study in a case-control subsample (N=274) using Illumina's Human-1 BeadChip. The phenotype definition for the cases of treatment-emergent suicidal ideation was the same as previously reported by this group. However, control group participants were selected on the basis of a minimum of three visits, a baseline QIDS-C total score ≥10, adherence to medication, recurrent depression, no history of suicide attempts, and no report of suicidal thoughts throughout citalopram treatment as measured by the QIDS-C and QIDS-SR. Two more markers were implicated in treatment-emergent suicidal ideation: one residing in the PAPLN gene (rs11628713), which encodes papilin, a protoglycan-like sulfated glycoprotein, and another that showed a strong trend after permutation correction located in the IL28RA gene (rs10903034) that encodes an interleukin receptor ( 67 ). Although these markers offer promising results with some meaningful effect sizes, the findings are still not sufficient to warrant use of these markers in a clinical setting ( Table 3 ).
Sexual dysfunction
Sexual dysfunction is a common adverse event that is a major contributor to treatment nonadherence among patients treated with SSRIs. One study of STAR*D data derived phenotypes from self-reports of erectile dysfunction, decreased libido, or anorgasmia on the Patient-Rated Inventory of Side Effects. When a set-based test was used, SNPs in glutamatergic genes were associated with decreased libido (GRIA3 and GRIK2), with difficulty achieving orgasm (GRIA1), and with difficulty achieving erection (GRIN3A) ( 34 ) ( Table 3 ). These results will require replication in an independent sample.
Conclusions and future directions
Although several pharmacogenetics studies have been conducted using STAR*D data, there is still much work to be done. The initial genomewide association studies were performed with now-obsolete platforms that provide genomewide coverage but that do not have the accuracy, marker density, and comprehensive coverage of the newer platforms available today. Until we fully understand the mechanisms that regulate gene function, we should maximize the results we obtain from available samples not only with genotyping with denser coverage but also with techniques such as sequencing and gene expression.
Every study conducted with STAR*D data has attempted correction for multiple testing to avoid false positives. However, replication of findings in independent samples is important in increasing our confidence in the reported results. Achieving replication of results with small effects or low allele frequencies might require a sample at least as large and as uniformly treated and longitudinally assessed as the STAR*D cohort. Moreover, outcome predictors based on multiple markers require still larger samples. These new samples may require not only further clinical characterization but also functional data such as neuroimaging or other biological markers.
A consensus is needed on phenotype definitions, particularly of antidepressant outcome. Without such definitions, replication studies and meta-analyses are difficult to interpret. Although more complex and thorough definitions might impair statistical power in small studies, STAR*D has helped to identify important confounders, such as tolerability, in analyses of the SLC6A4 (serotonin transporter). Adequate phenotype definitions for pharmacogenetic studies of antidepressant outcome should emphasize sufficient severity of baseline symptoms (for example, a HAM-D score of ≥14), sufficient exposure to the drug (for example, four to six weeks or more of treatment with an antidepressant), and adherence and tolerability. Without these characteristics, phenotypes could be uninterpretable and outcome results difficult to replicate across cohorts.
STAR*D has yielded a number of leads for future pharmacogenetics studies. Although results have not yet translated into findings that are clinically useful, genetic association studies are providing significant insights into the pathophysiology of depression. In summary, the next several years should bring significant progress to the field of pharmacogenetics. Investigators will continue to identify markers and genes involved in antidepressant treatment outcome and adverse effects that will increase our understanding of the biology of mood disorders and may identify predictors of outcome that would result in more effective treatments, fewer adverse events, and a greatly reduced burden of mood disorders for patients and society.
Acknowledgments and disclosures
STAR*D is supported by federal funds from NIMH under contract N01 MH-90003 to the University of Texas-Southwestern Medical Center at Dallas (Dr. Rush, principal investigator). This study was funded by the Intramural Research Program of NIMH, National Institutes of Health (NIH), U.S. Department of Health and Human Services (DHHS), NIMH grant K23MH67060, a NARSAD Young Investigator/Sidney R. Baer Jr. Foundation Award, and a Bowman Family Foundation award. The content of this publication does not necessarily reflect the views or policies of DHHS nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Dr. Laje, Dr. Rush, and Dr. McMahon are listed as coinventors on a patent application filed by NIH that is based in part on the diagnostic technology described in this article. The authors are not involved in the negotiation and execution of this license, but under federal law NIH is required to pay them a portion of any royalties NIH receives under the license. The authors do not endorse any commercial use of the patent. Dr. Rush has been an advisory board member, consultant, or speaker for Advanced Neuromodulation Systems, AstraZeneca, Bristol-Myers Squibb, Cyberonics, Forest Pharmaceuticals, Gerson Lehman Group, GlaxoSmithKline, Jazz Pharmaceuticals, Magellan Health Services, Merck, Neuronetics, Novartis, Ono, Organon, Otsuka, Pam Labs, Pfizer, Trancept, and Wyeth. Dr. Perlis has received advisory or consulting fees from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Pfizer, and Proteus LLC; he has received speaking fees or honoraria from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, and Pfizer; and he has equity holdings and patents with Concordant Rater Systems, LLC.
1. Murray CJ, Lopez AD: Evidence-based health policy: lessons from the Global Burden of Disease Study. Science 274:740–743, 1996Google Scholar
2. Nierenberg AA: Predictors of response to antidepressants: general principles and clinical implications. Psychiatric Clinics of North America 26:345–352, 2003Google Scholar
3. Trivedi MH, Rush AJ, Wisniewski SR, et al: Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. American Journal of Psychiatry 163:28–40, 2006Google Scholar
4. Gibbons RD, Brown CH, Hur K, et al: Early evidence on the effects of regulators' suicidality warnings on SSRI prescriptions and suicide in children and adolescents. American Journal of Psychiatry 164:1356–1363, 2007Google Scholar
5. Swen JJ, Huizinga TW, Gelderblom H, et al: Translating pharmacogenomics: challenges on the road to the clinic. PLoS Medicine 4:e209, 2007Google Scholar
6. Rush AJ, Fava M, Wisniewski SR, et al: Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Controlled Clinical Trials 25:119–142, 2004Google Scholar
7. Ioannidis JP, Trikalinos TA, Ntzani EE, et al: Genetic associations in large versus small studies: an empirical assessment. Lancet 361:567–571, 2003Google Scholar
8. Laje G, Paddock S, Manji H, et al: Genetic markers of suicidal ideation emerging during citalopram treatment of major depression. American Journal of Psychiatry 164:1530–1538, 2007Google Scholar
9. Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics 155:945–959, 2000Google Scholar
10. Pritchard JK, Stephens M, Rosenberg NA, et al: Association mapping in structured populations. American Journal of Human Genetics 67:170–181, 2000Google Scholar
11. Price AL, Patterson NJ, Plenge RM, et al: Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38:904–909, 2006Google Scholar
12. Hamilton M: A rating scale for depression. Journal of Neurology, Neurosurgery, and Psychiatry 23:56–62, 1960Google Scholar
13. Hamilton M: Development of a rating scale for primary depressive illness. British Journal of Social and Clinical Psychology 6:278–296, 1967Google Scholar
14. Rush AJ, Trivedi MH, Ibrahim HM, et al: The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C) and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biological Psychiatry 54:573–583, 2003Google Scholar
15. Rush AJ, Carmody TJ, Reimitz PE: The Inventory of Depressive Symptomatology (IDS): clinician (IDS-C) and self-report (IDS-SR) ratings of depressive symptoms. International Journal of Methods in Psychiatric Research 9:45–59, 2000Google Scholar
16. Wisniewski SR, Rush AJ, Balasubramani GK, et al: Self-rated global measure of the frequency, intensity, and burden of side effects. Journal of Psychiatric Practice 12:71–79, 2006Google Scholar
17. Rush AJ, Bernstein IH, Trivedi MH, et al: An evaluation of the quick inventory of depressive symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression Trial report. Biological Psychiatry 59:493–501, 2006Google Scholar
18. Yates WR, Mitchell J, John RA, et al: Clinical features of depression in outpatients with and without co-occurring general medical conditions in STAR*D: confirmatory analysis. Primary Care Companion to the Journal of Clinical Psychiatry 9:7–15, 2007Google Scholar
19. Davis LL, Frazier E, Husain MM, et al: Substance use disorder comorbidity in major depressive disorder: a confirmatory analysis of the STAR*D cohort. American Journal on Addictions 15:278–285, 2006Google Scholar
20. Davis LL, Frazier EC, Gaynes BN, et al: Are depressed outpatients with and without a family history of substance use disorder different? A baseline analysis of the STAR*D cohort. Journal of Clinical Psychiatry 68:1931–1938, 2007Google Scholar
21. Lin EH, Von KM, Katon W, et al: The role of the primary care physician in patients' adherence to antidepressant therapy. Medical Care 33:67–74, 1995Google Scholar
22. Serretti A, Kato M, Kennedy JL: Pharmacogenetic studies in depression: a proposal for methodologic guidelines. Pharmacogenomics Journal 8:90–100, 2008Google Scholar
23. Newton-Howes G, Tyrer P, Johnson T: Personality disorder and the outcome of depression: meta-analysis of published studies. British Journal of Psychiatry 188:13–20, 2006Google Scholar
24. Kool S, Schoevers R, de Maat S, et al: Efficacy of pharmacotherapy in depressed patients with and without personality disorders: a systematic review and meta-analysis. Journal of Affective Disorders 88:269–278, 2005Google Scholar
25. Russell JM, Kornstein SG, Shea MT, et al: Chronic depression and comorbid personality disorders: response to sertraline versus imipramine. Journal of Clinical Psychiatry 64:554–561, 2003Google Scholar
26. Hirschfeld RM, Klerman GL, Clayton PJ, et al: Assessing personality: effects of the depressive state on trait measurement. American Journal of Psychiatry 140:695–699, 1983Google Scholar
27. McGrath PJ, Stewart JW, Petkova E, et al: Predictors of relapse during fluoxetine continuation or maintenance treatment of major depression. Journal of Clinical Psychiatry 61:518–524, 2000Google Scholar
28. Quitkin FM, Rabkin JD, Markowitz JM, et al: Use of pattern analysis to identify true drug response: a replication. Archives of General Psychiatry 44:259–264, 1987Google Scholar
29. Stewart JW, Quitkin FM, McGrath PJ, et al: Use of pattern analysis to predict differential relapse of remitted patients with major depression during 1 year of treatment with fluoxetine or placebo. Archives of General Psychiatry 55:334–343, 1998Google Scholar
30. McGrath PJ, Stewart JW, Quitkin FM, et al: Predictors of relapse in a prospective study of fluoxetine treatment of major depression. American Journal of Psychiatry 163:1542–1548, 2006Google Scholar
31. Perlis RH, Moorjani P, Fagerness J, et al: Pharmacogenetic analysis of genes implicated in rodent models of antidepressant response: association of TREK1 and treatment resistance in the STAR*D study. Neuropsychopharmacology 33:2810–2819, 2008Google Scholar
32. McMahon FJ, Buervenich S, Charney D, et al: Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. American Journal of Human Genetics 78:804–814, 2006Google Scholar
33. Furr LA: Perceptions of genetics research as harmful to society: differences among samples of African-Americans and European-Americans. Genetic Testing 6:25–30, 2002Google Scholar
34. Perlis RH, Laje G, Smoller JW, et al: Genetic and clinical predictors of sexual dysfunction in citalopram-treated depressed patients. Neuropsychopharmacology 34:1819–1828, 2009Google Scholar
35. Paddock S, Laje G, Charney D, et al: Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. American Journal of Psychiatry 164:1181–1188, 2007Google Scholar
36. Hu XZ, Rush AJ, Charney D, et al: Association between a functional serotonin transporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Archives of General Psychiatry 64:783–792, 2007Google Scholar
37. Mrazek DA, Rush AJ, Biernacka JM, et al: SLC6A4 variation and citalopram response. American Journal of Medical Genetics Part B, Neuropsychiatric Genetics 150:341–351, 2008Google Scholar
38. Lekman M, Laje G, Charney D, et al: The FKBP5-gene in depression and treatment response: an association study in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) cohort. Biological Psychiatry 63:1103–1110, 2008Google Scholar
39. Cabanero M, Laje G, Detera-Wadleigh S, et al: Association study of phosphodiesterase genes in the Sequenced Treatment Alternatives to Relieve Depression sample. Pharmacogenetics and Genomics 19:235–238, 2009Google Scholar
40. Teranishi KS, Slager SL, Garriock H, et al: Variants in PDE11A and PDE1A are not associated with citalopram response. Molecular Psychiatry 12:1061–1063, 2007Google Scholar
41. Domschke K, Lawford B, Laje G, et al: Brain-derived neurotrophic factor (BDNF) gene: no major impact on antidepressant treatment response. International Journal of Neuropsychopharmacology 23:1–9, 2009Google Scholar
42. Peters EJ, Slager SL, Kraft JB, et al: Pharmacokinetic genes do not influence response or tolerance to citalopram in the STAR*D sample. PLoS One 3:e1872, 2008Google Scholar
43. Rasmussen HB, Werge TM: Novel procedure for genotyping of the human serotonin transporter gene-linked polymorphic region (5-HTTLPR)—a region with a high level of allele diversity. Psychiatric Genetics 17:287–291, 2007Google Scholar
44. Serretti A, Kato M, De Ronchi D, et al: Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Molecular Psychiatry 12:247–257, 2007Google Scholar
45. Kraft JB, Peters EJ, Slager SL, et al: Analysis of association between the serotonin transporter and antidepressant response in a large clinical sample. Biological Psychiatry 61:734–742, 2007Google Scholar
46. Binder EB, Salyakina D, Lichtner P, et al: Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nature Genetics 36:1319–1325, 2004Google Scholar
47. Kirchheiner J, Lorch R, Lebedeva E, et al: Genetic variants in FKBP5 affecting response to antidepressant drug treatment. Pharmacogenomics 9:841–846, 2008Google Scholar
48. Tsai SJ, Hong CJ, Chen TJ, et al: Lack of supporting evidence for a genetic association of the FKBP5 polymorphism and response to antidepressant treatment. American Journal of Medical Genetics Part B, Neuropsychiatric Genetics 144:1097–1098, 2007Google Scholar
49. Wong ML, Whelan F, Deloukas P, et al: Phosphodiesterase genes are associated with susceptibility to major depression and antidepressant treatment response. Proceedings of the National Academy of Sciences of the United States of America 103:15124–15129, 2006Google Scholar
50. Nair A, Vaidya VA: Cyclic AMP response element binding protein and brain-derived neurotrophic factor: molecules that modulate our mood? Journal of Bioscience 31:423–434, 2006Google Scholar
51. Green E, Craddock N: Brain-derived neurotrophic factor as a potential risk locus for bipolar disorder: evidence, limitations, and implications. Current Psychiatry Reports 469–476, 2003Google Scholar
52. Saarelainen T, Hendolin P, Lucas G, et al: Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. Journal of Neuroscience 23:349–357, 2003Google Scholar
53. Egan MF, Kojima M, Callicott JH, et al: The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112:257–269, 2003Google Scholar
54. Ventriglia M, Bocchio Chiavetto L, Benussi L, et al: Association between the BDNF 196 A/G polymorphism and sporadic Alzheimer's disease. Molecular Psychiatry 7:136–137, 2002Google Scholar
55. Hwang JP, Tsai SJ, Hong CJ, et al: The Val66Met polymorphism of the brain-derived neurotrophic-factor gene is associated with geriatric depression. Neurobiology of Aging 27:1834–1837, 2006Google Scholar
56. Ribeiro L, Busnello JV, Cantor RM, et al: The brain-derived neurotrophic factor rs6265 (Val66Met) polymorphism and depression in Mexican-Americans. Neuroreport 18:1291–1239, 2007Google Scholar
57. Schumacher J, Jamra RA, Becker T, et al: Evidence for a relationship between genetic variants at the brain-derived neurotrophic factor (BDNF) locus and major depression. Biological Psychiatry 58:307–314, 2005Google Scholar
58. Hong CJ, Huo SJ, Yen FC, et al: Association study of a brain-derived neurotrophic-factor genetic polymorphism and mood disorders, age of onset and suicidal behavior. Neuropsychobiology 48:186–189, 2003Google Scholar
59. Tsai SJ, Cheng CY, Yu YW, et al: Association study of a brain-derived neurotrophic-factor genetic polymorphism and major depressive disorders, symptomatology, and antidepressant response. American Journal of Medical Genetics Part B, Neuropsychiatric Genetics 123:19–22, 2003Google Scholar
60. Chen L, Lawlor DA, Lewis SJ, et al: Genetic association study of BDNF in depression: finding from two cohort studies and a meta-analysis. American Journal of Medical Genetics Part B, Neuropsychiatric Genetics 147B:814–821, 2008Google Scholar
61. Choi MJ, Kang RH, Lim SW, et al: Brain-derived neurotrophic factor gene polymorphism (Val66Met) and citalopram response in major depressive disorder. Brain Research 1118:176–182, 2006Google Scholar
62. Anttila S, Huuhka K, Huuhka M, et al: Interaction between 5-HT1A and BDNF genotypes increases the risk of treatment-resistant depression. Journal of Neural Transmission 114:1065–1068, 2007Google Scholar
63. Peters EJ, Slager SL, Jenkins GD, et al: Resequencing of serotonin-related genes and association of tagging SNPs to citalopram response. Pharmacogenetics and Genomics 19:1–10, 2009Google Scholar
64. Antidepressant Use in Children, Adolescents and Adults. Silver Spring, Md, US Food and Drug Administration. Available at fda.gov/drugs/drugsafety/informationbydrugclass Google Scholar
65. Perlis RH, Purcell S, Fava M, et al: Association between treatment-emergent suicidal ideation with citalopram and polymorphisms near cyclic adenosine monophosphate response element binding protein in the STAR*D study. Archives of General Psychiatry 64:689–697, 2007Google Scholar
66. Menke A, Lucae S, Kloiber S, et al: Genetic markers within glutamate receptors associated with antidepressant treatment-emergent suicidal ideation. American Journal of Psychiatry 165:917–918, 2008Google Scholar
67. Laje G, Allen AS, Akula N, et al: Genome-wide association study of suicidal ideation emerging during citalopram treatment of depressed outpatients. Pharmacogenetics and Genomics, 19:666–674, 2009Google Scholar

